

概述
下一代测序(NGS)技术研究整个基因组(全基因组测序)或目标区域(例如全外显子组测序)的基因组变异。它可用来自胎儿/个体的不同类型样本的DNA进行分析,如绒毛,羊水,脐带血,唾液和外周血等。与G显带染色体核型分析技术和染色体微阵列分析(CMA)技术相比,NGS在产前/产后/生殖基因检测中能够提高遗传性疾病的诊断率(PMID:32451733)。
当前,我们提供三种不同的基于NGS的基因组测序服务,包括:
FetalSeq
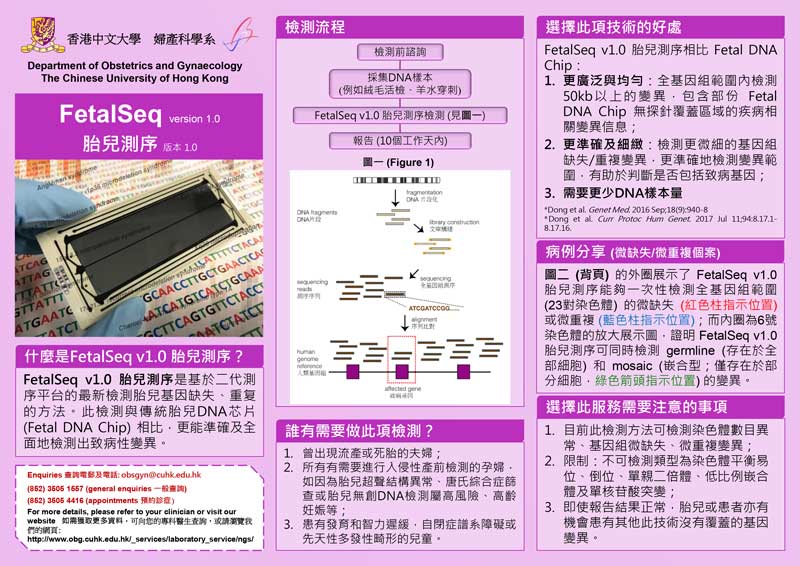
FetalSeq是一项基于low-pass(低覆盖率但高通量)原理的全基因组测序(LP-WGS)技术,它采用内部开发的生物信息学分析流程检测有临床意义的拷贝数变异(CNVs)和染色体非整倍体。该技术是由香港中文大学开发并验证的。FetalSeq在不同应用中提供全面的基因组研究,包括流产,针对高危妊娠的侵入性产前诊断(如胎儿超声异常和/或其他适应症等),以及发育迟缓,智力障碍,自闭症谱系障碍或先天性异常等产后病例(PMIDs:26820068,28696555,31447483和32451733)。FetalSeq可检测几乎所有已知的微缺失和微重复综合征,以及大于50kb的拷贝数变异。与CNV-seq相比,它具有更高的全基因组分辨率(PMID:24998187)。
方法:提取样本中的基因组DNA(50-100ng)进行非靶向富集构建文库。利用下一代测序平台(包括NovaSeq,HiSeq X Ten和BGISEQ-500 / MGISEQ-2000等)对样品进行测序。常规测序深度约为0.25×(约1千5百万条测序片段,单末端测序片段长度为50bp)。使用Burrows-Wheeler比对工具(BWA)(PMID:19451168)将测序片段与人类参考序列(GRCh37 / hg19)进行比对,并根据比对的坐标划分入50kb大小的滑动窗口(增量为5kb)。经过GC校正和基于人群的归一化之后,通过内部的生物信息分析流程(覆盖率增量)进行CNV检测。根据美国医学遗传学和基因组学协会的指南,参考医学文献和在线数据库进行数据解释。
局限性:FetalSeq不能排除人类基因组中所有小的染色体异常(<50kb),拷贝数多态性和染色体重排(PMID:15286789)。FetalSeq不能检测三倍体,单亲二倍体,低水平嵌合(<30%),三核苷酸重复扩增,平衡的结构变异,插入缺失或点突变。
报告周期(TAT):10个工作日
样本要求:
| 样品类型 | 量 |
|---|---|
| 血液 | 2-3 ml (EDTA瓶中) |
| 绒毛 | 5 mg绒毛(运送培养基中) |
| 羊水 | > 10 ml(无菌容器中) |
| 胎盘组织 | 至少0.5ug 最低浓度为30ng / uL;A260 / A280〜1.8 |

ChromoSeq
ChromoSeq是一项基于mate-pair测序原理(DNA片段大小约5kb)的low-pass 全基因组测序(LP-WGS),是由香港中文大学开发和验证的遗传学检测,有内部开发的生物信息学分析流程,可应用于产前和产后的遗传诊断,以及复发性流产夫妇的检测(PMIDs:24610732,29095815,29364520,31173071和31679651)。除了染色体非整倍体(例如唐氏综合征)和拷贝数变异(例如DiGeorge综合征)外,ChromoSeq还可检测其他染色体结构重排,例如平衡易位,倒位,插入和复杂重排。此外,它还能鉴别杂合性缺失和单亲二倍体。某些印记基因区域的杂合性缺失或单亲二倍体会引起疾病(例如Beckwith Wiedemann综合征,Russell-Silver综合征)。
ChromoSeq检测(1)几乎所有已知的微缺失和微重复综合征以及大于50kb且有临床意义的拷贝数变异;(2)结构重排,例如平衡易位,倒位,插入和复杂的结构重排;(3)分辨率为5Mb的AOH。ChromoSeq是FetalSeq的增强版。
方法:从样品中提取基因组DNA(2ug),使用大片段的DNA(〜5kb,PMID:31173071)基于mate-pair原理进行内部文库构建(CP-AL)。该文库在NovaSeq,HiSeq X Ten和MGISEQ-2000等下一代测序平台上进行测序。常规测序深度约为3×(约5000万对测序片段,双末端测序长度100bp)。使用Burrows-Wheeler比对工具(BWA)(PMID:19451168)将测序片段与参考序列(GRCh37 / hg19)进行比对。(1)CNV检测:根据比对的坐标,将测序片段划分入50kb大小的滑动窗口(增量为5kb)。经过GC校正和基于人群的归一化之后,通过内部流程(覆盖率递增)进行CNV检测,并根据美国医学遗传学和基因组学协会的指南并参考医学文献和在线数据库进行解释。(2)结构重排检测:基于以下四个步骤对唯一比对序列进一步处理:事件聚类,系统错误过滤,随机错误过滤和比对定位。(3)AOH检测:通过分析mpileup文件(Samtools)进行基因分型。杂合性SNVs降低和纯合性SNVs增加的区域(>5Mb)被鉴定为AOH区域。
局限性:ChromoSeq并不排除所有小的染色体异常(<50Kb),拷贝数多态性。ChromoSeq结果正常不能排除存在三倍体,三核苷酸重复扩增,低水平嵌合(<30%),插入缺失或点突变。
报告周期(TAT):3周
样本要求:请致电(852)3505 1557与我们的实验室联系!
常问问题
Q1。由于ChromoSeq可以检测到平衡易位,因此不再需要寄送父母血液样本进行核型分析,是吗?
回答:是的,ChromoSeq在鉴定各种复杂的染色体结构变异(包括平衡易位,倒位以及微缺失/重复)方面优于核型分析。但是,ChromoSeq的局限性在于无法检测罗伯逊易位。因此,如果在妊娠物中检测到端着丝粒染色体(13、14、15、21、22)反复发生三体现象,仍需进行父母染色体核型分析以排除罗伯逊易位。
Q2。对于复发性流产患者,我是否应该将流产物(POG)先进行ChromoSeq检测(无需送父母样本,对吗?)?
回答:我们希望父母样本和POG样本一起进行ChromoSeq检测。当然,我们可以首先检测POG是否存在非整倍体,全基因组范围的致病性拷贝数变异(CNV),结构重排(SV)和杂合性缺失(AOH,例如单亲二倍体,UPD)。ChromoSeq采用家系分析(与父母样品一起检测)的目的是解释遗传自父母的有临床意义的变异。与CMA或FetalSeq类似,ChromoSeq可以识别出先证者或胎儿基因组中的隐匿变异(CNV,SV和AOH),需要这些变异的亲本遗传信息用于进一步的临床解释。
Q3。进行ChromoSeq +核型分析有什么意义?
响应:如Q1所述,ChromoSeq的局限性在于无法检测罗伯逊易位。为了全面鉴定父母样本的遗传背景,因此需要进行两项检测。这也是我们提供较低价格套餐的原因。
Q4。如果我给您寄送了妊娠物样本,但您的团队找不到绒毛,那费用怎么算?回应:我们只会收取500元港币的手续费。但是,如果您还订购了核型分析,则将另外收取1,000元港币的费用。


GenomSeq
GenomSeq是最全面的基因组检测之一,可在更高的测序深度下对几乎全部基因组进行测序。基于下一代测序技术和我们内部的生物信息学分析流程,GenomSeq可全面识别基因组变异,包括单核苷酸变异(SNV),拷贝数变异(CNV),结构重排以及杂合性缺失(AOH)。该方法由香港中文大学开发并验证。GenomSeq的应用包括产前和产后遗传学诊断(PMID:31475041))。该技术检测(1)具有临床意义的单核苷酸变异;(2)几乎所有已知的微缺失和微重复综合症,以及大于50kb且有临床意义的拷贝数变异;(3)结构重排,如平衡易位,倒位,插入和复杂的结构重排;(4)分辨率为5Mb的AOH。除了ChromoSeq可以检测到的遗传异常外,GenomSeq还能鉴定致病性单核苷酸变异,这些变异会通过破坏基因的正常功能而导致遗传性疾病(例如Beta地中海贫血,Noonan综合征)。GenomSeq是ChromoSeq的增强版。
方法:将样品中提取的基因组DNA(500-1000ng)采用非靶向富集的PCR-free方法进行文库构建,并在NovaSeq,HiSeq X Ten和BGISEQ-500 / MGISEQ-2000等平台进行测序。常规测序深度约为30×(约4.5亿对测序片段,双末端测序片段大小为100bp)。使用Burrows-Wheeler比对工具(BWA)(PMID:19451168)将测序片段与参考序列(GRCh37 / hg19)进行比对。(1)SNV检测:采用HaplotypeCaller(GATK)进行分析,并用ANNOVAR结合不同的参考数据库进行注释(PMID: 20601685)。根据美国医学遗传学和基因组学协会的指南,参考医学文献和在线数据库对SNV进行解释。(2)CNV检测:根据比对的坐标,将测序片段划分入50kb大小的滑动窗口(增量为5kb)。经过GC校正和基于人群的归一化之后,通过内部流程(覆盖率递增)进行CNV检测,并根据美国医学遗传学和基因组学协会的指南并参考医学文献和在线数据库进行解释。(3)结构重排检测:基于以下四个步骤对唯一比对序列进一步处理:事件聚类,系统错误过滤,随机错误过滤和比对定位。(4)AOH检测:通过分析mpileup文件(Samtools)进行基因分型。杂合性SNVs降低和纯合性SNVs增加的区域(>5Mb)被鉴定为AOH区域。
局限性:GenomSeq并不能排除所有小的染色体异常,例如三核苷酸重复扩增,三倍体和低水平嵌合(<30%)。
报告周期(TAT):即将推出!
样本要求:即将推出!