

概述
下一代測序(NGS)技術研究整個基因組(全基因組測序)或目的地區域(例如全外顯子組測序)的基因組變異。它可用來自胎兒/個體的不同類型樣本的DNA進行分析,如絨毛,羊水,臍帶血,唾液和外周血等。與G顯帶染色體核型分析技術和染色體微陣列分析(CMA)技術相比,NGS在產前/產後/生殖基因檢測中能夠提高遺傳性疾病的診斷率(PMID:32451733)。
當前,我們提供三種不同的基於NGS的基因組測序服務,包括:
FetalSeq
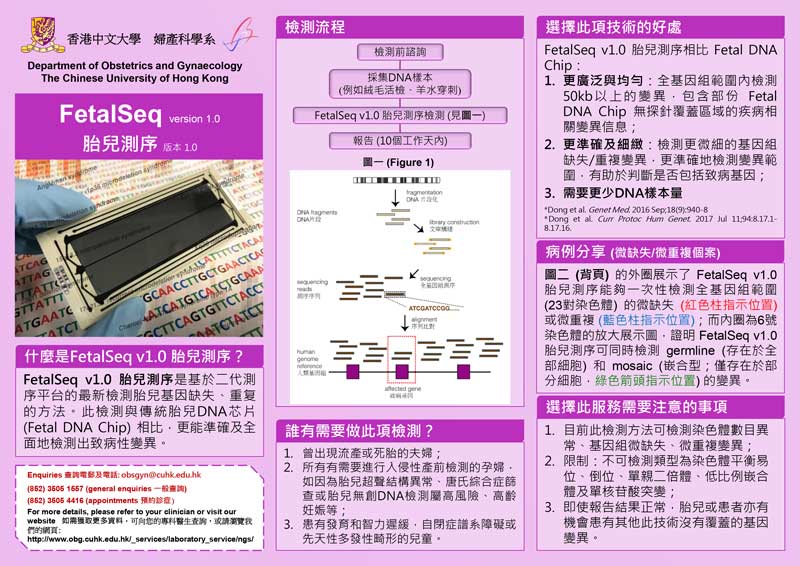
FetalSeq是一項基於low-pass(低覆蓋率但高通量)原理的全基因組測序(LP-WGS)技術,它採用內部開發的生物資訊學分析流程檢測有臨床意義的拷貝數變異(CNVs)和染色體非整倍體。該技術是由香港中文大學開發並驗證的。FetalSeq在不同應用中提供全面的基因組研究,包括流產,針對高危妊娠的侵入性產前診斷(如胎兒超聲異常和/或其他適應症等),以及發育遲緩,智力障礙,自閉症譜系障礙或先天性異常等產後病例(PMIDs:26820068,28696555,31447483和32451733)。FetalSeq可檢測幾乎所有已知的微缺失和微重複綜合征,以及大於50kb的拷貝數變異。與CNV-seq相比,它具有更高的全基因組解析度(PMID:24998187)。
方法:提取樣本中的基因組DNA(50-100ng)進行非靶向富集構建文庫。利用下一代測序平臺(包括NovaSeq,HiSeq X Ten和BGISEQ-500 / MGISEQ-2000等)對樣品進行測序。常規測序深度約為0.25×(約1千5百萬條測序片段,單末端測序片段長度為50bp)。使用Burrows-Wheeler比對工具(BWA)(PMID:19451168)將測序片段與人類參考序列(GRCh37 / hg19)進行比對,並根據比對的座標劃分入50kb大小的滑動窗口(增量為5kb)。經過GC校正和基於人群的歸一化之後,通過內部的生物資訊分析流程(覆蓋率增量)進行CNV檢測。根據美國醫學遺傳學和基因組學協會的指南,參考醫學文獻和線上資料庫進行資料解釋。
局限性:FetalSeq不能排除人類基因組中所有小的染色體異常(<50kb),拷貝數多態性和染色體重排(PMID:15286789)。FetalSeq不能檢測三倍體,單親二倍體,低水準嵌合(<30%),三核苷酸重複擴增,平衡的結構變異,插入缺失或點突變。
報告週期(TAT):10個工作日
樣本要求:
| 樣品類型 | 量 |
|---|---|
| 血液 | 2-3 ml (EDTA瓶中) |
| 絨毛 | 5 mg絨毛(運送培養基中) |
| 羊水 | > 10 ml(無菌容器中) |
| 胎盤組織 | 至少0.5ug 最低濃度為30ng / uL;A260 / A280〜1.8 |

ChromoSeq
ChromoSeq是一項基於mate-pair測序原理(DNA片段大小約5kb)的low-pass 全基因組測序(LP-WGS),是由香港中文大學開發和驗證的遺傳學檢測,有內部開發的生物資訊學分析流程,可應用於產前和產後的遺傳診斷,以及復發性流產夫婦的檢測(PMIDs:24610732,29095815,29364520,31173071和31679651)。除了染色體非整倍體(例如唐氏綜合征)和拷貝數變異(例如DiGeorge綜合征)外,ChromoSeq還可檢測其他染色體結構重排,例如平衡易位,倒位,插入和複雜重排。此外,它還能鑒別雜合性缺失和單親二倍體。某些印記基因區域的雜合性缺失或單親二倍體會引起疾病(例如Beckwith Wiedemann綜合征,Russell-Silver綜合征)。
ChromoSeq檢測(1)幾乎所有已知的微缺失和微重複綜合征以及大於50kb且有臨床意義的拷貝數變異;(2)結構重排,例如平衡易位,倒位,插入和複雜的結構重排;(3)解析度為5Mb的AOH。ChromoSeq是FetalSeq的增強版。
方法:從樣品中提取基因組DNA(2ug),使用大片段的DNA(〜5kb,PMID:31173071)基於mate-pair原理進行內部文庫構建(CP-AL)。該文庫在NovaSeq,HiSeq X Ten和MGISEQ-2000等下一代測序平臺上進行測序。常規測序深度約為3×(約5000萬對測序片段,雙末端測序長度100bp)。使用Burrows-Wheeler比對工具(BWA)(PMID:19451168)將測序片段與參考序列(GRCh37 / hg19)進行比對。(1)CNV檢測:根據比對的座標,將測序片段劃分入50kb大小的滑動視窗(增量為5kb)。經過GC校正和基於人群的歸一化之後,通過內部流程(覆蓋率遞增)進行CNV檢測,並根據美國醫學遺傳學和基因組學協會的指南並參考醫學文獻和線上資料庫進行解釋。(2)結構重排檢測:基於以下四個步驟對唯一比對序列進一步處理:事件聚類,系統錯誤過濾,隨機錯誤過濾和比對定位。(3)AOH檢測:通過分析mpileup檔(Samtools)進行基因分型。雜合性SNVs降低和純合性SNVs增加的區域(>5Mb)被鑒定為AOH區域。
局限性:ChromoSeq並不排除所有小的染色體異常(<50Kb),拷貝數多態性。ChromoSeq結果正常不能排除存在三倍體,三核苷酸重複擴增,低水準嵌合(<30%),插入缺失或點突變。
報告週期(TAT):3周
樣本要求:請致電(852)3505 1557與我們的實驗室聯繫!
常問問題
Q1。由於ChromoSeq可以檢測到平衡易位,因此不再需要寄送父母血液樣本進行核型分析,是嗎?
回答:是的,ChromoSeq在鑒定各種複雜的染色體結構變異(包括平衡易位,倒位以及微缺失/重複)方面優於核型分析。但是,ChromoSeq的局限性在於無法檢測羅伯遜易位。因此,如果在妊娠物中檢測到端著絲粒染色體(13、14、15、21、22)反復發生三體現象,仍需進行父母染色體核型分析以排除羅伯遜易位。
Q2。對於復發性流產患者,我是否應該將流產物(POG)先進行ChromoSeq檢測(無需送父母樣本,對嗎?)?
回答:我們希望父母樣本和POG樣本一起進行ChromoSeq檢測。當然,我們可以首先檢測POG是否存在非整倍體,全基因組範圍的致病性拷貝數變異(CNV),結構重排(SV)和雜合性缺失(AOH,例如單親二倍體,UPD)。ChromoSeq採用家系分析(與父母樣品一起檢測)的目的是解釋遺傳自父母的有臨床意義的變異。與CMA或FetalSeq類似,ChromoSeq可以識別出先證者或胎兒基因組中的隱匿變異(CNV,SV和AOH),需要這些變異的親本遺傳信息用於進一步的臨床解釋。
Q3。進行ChromoSeq +核型分析有什麼意義?
回應:如Q1所述,ChromoSeq的局限性在於無法檢測羅伯遜易位。為了全面鑒定父母樣本的遺傳背景,因此需要進行兩項檢測。這也是我們提供較低價格套餐的原因。
Q4。如果我給您寄送了妊娠物樣本,但您的團隊找不到絨毛,那費用怎麼算?回應:我們只會收取500元港幣的手續費。但是,如果您還訂購了核型分析,則將另外收取1,000元港幣的費用。


GenomSeq
GenomSeq是最全面的基因組檢測之一,可在更高的測序深度下對幾乎全部基因組進行測序。基於下一代測序技術和我們內部的生物資訊學分析流程,GenomSeq可全面識別基因組變異,包括單核苷酸變異(SNV),拷貝數變異(CNV),結構重排以及雜合性缺失(AOH)。該方法由香港中文大學開發並驗證。GenomSeq的應用包括產前和產後遺傳學診斷(PMID:31475041))。該技術檢測(1)具有臨床意義的單核苷酸變異;(2)幾乎所有已知的微缺失和微重複綜合症,以及大於50kb且有臨床意義的拷貝數變異;(3)結構重排,如平衡易位,倒位,插入和複雜的結構重排;(4)解析度為5Mb的AOH。除了ChromoSeq可以檢測到的遺傳異常外,GenomSeq還能鑒定致病性單核苷酸變異,這些變異會通過破壞基因的正常功能而導致遺傳性疾病(例如Beta地中海貧血,Noonan綜合征)。GenomSeq是ChromoSeq的增強版。
方法:將樣品中提取的基因組DNA(500-1000ng)採用非靶向富集的PCR-free方法進行文庫構建,並在NovaSeq,HiSeq X Ten和BGISEQ-500 / MGISEQ-2000等平臺進行測序。常規測序深度約為30×(約4.5億對測序片段,雙末端測序片段大小為100bp)。使用Burrows-Wheeler比對工具(BWA)(PMID:19451168)將測序片段與參考序列(GRCh37 / hg19)進行比對。(1)SNV檢測:採用HaplotypeCaller(GATK)進行分析,並用ANNOVAR結合不同的參考資料庫進行注釋(PMID: 20601685)。根據美國醫學遺傳學和基因組學協會的指南,參考醫學文獻和線上資料庫對SNV進行解釋。(2)CNV檢測:根據比對的座標,將測序片段劃分入50kb大小的滑動視窗(增量為5kb)。經過GC校正和基於人群的歸一化之後,通過內部流程(覆蓋率遞增)進行CNV檢測,並根據美國醫學遺傳學和基因組學協會的指南並參考醫學文獻和線上資料庫進行解釋。(3)結構重排檢測:基於以下四個步驟對唯一比對序列進一步處理:事件聚類,系統錯誤過濾,隨機錯誤過濾和比對定位。(4)AOH檢測:通過分析mpileup檔(Samtools)進行基因分型。雜合性SNVs降低和純合性SNVs增加的區域(>5Mb)被鑒定為AOH區域。
局限性:GenomSeq並不能排除所有小的染色體異常,例如三核苷酸重複擴增,三倍體和低水準嵌合(<30%)。
報告週期(TAT):即將推出!
樣本要求:即將推出!